... in which we get to know something about the work our hero does.
Bioinformatics?! WTF?
is the application of computer science to biological sciences. Basically, this means: data analysis. When lab people happily torture bacteria, squeeze out yeasts and puree mouse embryos, they generate data by the terabytes about genome sequences, RNA levels, protein abundance, small molecule concentrations and a lot more. To manage and analyze all that data, you need a lot of nerds with a basic understanding of what's happening inside a cell. They develop algorithms, find and apply statistical techniques and build databases, and if you're lucky, make sense out of your raw numbers.
But Bioinformatics has become a science in its own right. In Computational Systems Biology, we try to model living systems, or at least parts of them, like cellular subsystems, like metabolism. This is where I ended up (or started recently). By building a mathematical model of the processes inside a cell and testing that model with different parameters, we can find out what's happening under different circumstances, like changing nutrition, environmental stress or disruptions inside the cell without spending days and nights in the lab. This can be directly useful, e.g. for finding pills that maximally harm infectious bacteria, or engineer microorganisms that produce those pills or biofuels, but it also generates important hypotheses about the way life works and deals with all kinds of environments (which is something life is good at).
Metabolism is the chemistry of the cell: Incoming molecules (food) is broken down, yielding energy and building blocks (catabolism). From these blocks, all cellular components like DNA, proteins, membranes are built (anabolism). All this work is done by enzymes, which catalyze chemical reactions. These form a huge network, where the output of one reaction is the input for a multitude of others. The flow of molecules through that network determines what the cell can and will do. By understanding that network, its structure and its dynamics, we can redesign such networks to our advantage. The goal is to engineer microorganisms that can build plastic, biofuels, pharmaceuticals, out of organic material. Or even out of carbon dioxide using the sun as energy source (no more oil needed!). Or get rid of our toxic waste, using it as yummy food leaving nothing more than rainbows smelling like unicorn farts. The ultimate goal however is to design such networks from scratch, building tailor-made organisms[*].
[*] The last sentence refers to Synthetic Biology, which is still a little futuristic, given that we hardly understand what's going on in your ordinary garden variety E. coli, which has been studied to death and back for decades now. Also, nobody knows how unicorn farts smell.
Abstract
My interests focus on systems biology, especially all things metabolic. Analyzing structures and dynamics of complex biological networks and the application of graph theory and statistical methods to molecular biology data form the core of my PhD research.
The possibilities and promises of synthetic biology are quite fascinating. In particular, I am interested in creating a metabolic pathway planner, which will provide a blueprint for tailor-made microorganisms for particular tasks like biofuel and drug production or toxic waste disposal utilizing readily available food sources.
Basics
Atom mapping
Actually the topic of my diploma thesis, forms the basis for much of the work I am doing now. Given a chemical reaction equation with substrate and product molecules, which atom from the substrates corresponds to which atom in the products? The atom mapping is directly related to the reaction mechanism, since this determines which bonds are broken, formed or changed during that reaction (and vice versa). It is also an important question for determining mass flux for reactions and by extension in reaction networks. Where do the carbon atoms of a sugar molecule ultimately end up in the organism?
Based on graph representations of the molecular structure, we try to find isomorphic subgraphs of the substrates and products by iteratively fragmenting the molecules. Once we have found isomorphic fragments, the nodes (atoms) can be mapped to each other and back to the original molecule graphs. A greedy heuristic that maximizes the subgraph isomorphisms of the fragments produces results fast and is accurate for about ~91 % of enzymatic reactions in the KEGG database.
A web interface for the reaction mapper can be found at https://rna.tbi.univie.ac.at/cgi-bin/mapreaction.cgi. Publication is in progress (and has been for quite a while).
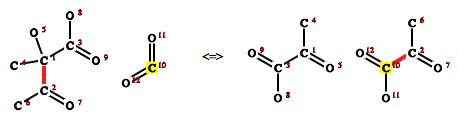
Above is a sample output from the web server. Atoms have been relabelled to match atom ids on the left side, broken (left) and formed (right) bonds are highlighted. One atom map pair is highlighted in yellow.
Graph grammars
When we represent molecules as graphs with atoms as nodes and the bonds between these atoms as edges, a chemical reaction on that molecule can then be seen as a graph rewrite operation: edges are added and/or removed (bonds broken and formed). A rewrite rule consists of three parts: left, context and right. The left part contains the edges that are removed, the right part of added edges (in chemical reactions, due to the law of mass conservation, no nodes (atoms) are created or destroyed). The context defines the applicability of the rule: If it is a (sub)graph of a candidate graph, the rule can transform this graph.
Graph grammars are a good model of enzymatic reactions. The context defines the substrate specificity of an enzyme. A comprehensive set of rewrite rules can recreate a reaction network computationally that looks and behaves like cellular metabolism. An efficient atom mapping algorithm can derive these rewrite rules from metabolic databases, such as KEGG or MetaCyc. Each rewrite rule then represents an enzyme and its activity. A graph grammar based artificial chemistry is constructive, i.e. it can create molecules that are not pre-defined. Thus, we can form hypotheses about parts of metabolic networks that have not been explored before, because we can find novel pathways and metabolites.
The subgraph isomorphism is a good approximation of the promiscuous behavior various enzymes exhibit, where there is catalytic activity on a broad variety of substrates, often with lower efficiency. This promiscuity results in a shadow metabolism that eludes lab analysis due to its low activity, but is quite important for understanding the behavior of cells during perturbations or changing conditions. Current hypotheses point to a scenario where gaps in the reaction network are filled with this shadow activity and enzymes are recruited from different parts of the network to compensate for missing enzymes or temporary substrate abundance. This might be important for the understanding and analysis of knock-out experiments or drug efficiency and can play an important role in metabolic engineering.
Enzyme clustering
A comprehensive set of rewrite rules defines the "synthesis space" of an organism, its chemical abilities to handle feed molecules, possible transformations and therefore the molecules it can produce. Many enzymes catalyze similar reactions on different substrates, or belong to a superclass of reactions that define the same basic transformation. In order to reduce the amount of rewrite rules needed to represent the metabolic activity of a cell, we develop a method to find classes of rewrite rules and derive super-rules that represent that class. These rules should be able to handle all the transformations while being as substrate specific as possible.
Since graph rewrite rules can themselves be seen as graphs, we define a graph distance measure based on a weighted maximum common subgraph of two rules. The subgraph edges are weighted according to their distance to the reaction sites, based on the hypothesis that context gets less important away from the active center of the enzyme. At the moment, this is implemented as a simple alignment of two graphs. An improved distance measure would also take into account various substitution probabilities for atoms, for example oxygen and sulphur exhibit more similar chemistry than oxygen and carbon.
Traditional clustering algorithms (hierarchical clustering) are then used to form enzyme clusters that contain enzymes with similar mechanisms. From all the rules in a cluster, consensus rules are built by graph alignments. This reduces the set of over 3000 enzymatic reactions in the KEGG database to approximately 400 rules that span the whole chemistry of life.
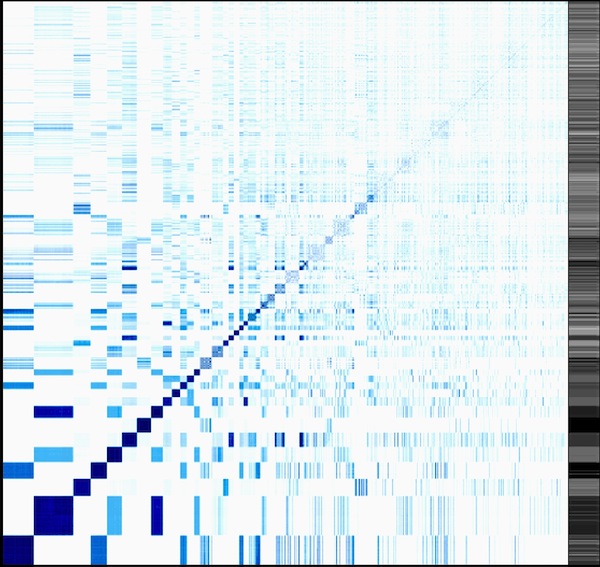
Preliminary results: Heatmap comparing the found clusters. A comparison with the traditional Enzyme Commission (EC) classification system can be found at https://www.tbi.univie.ac.at/~hekker/echeat/echeat.xhtml. Also take a look at Visual things#Enzyme clustering.
Thesis projects
Pathway planner
::::work in progress::::
A graph grammar based artificial chemistry will be used to predict novel pathways for the production of desired molecules like biofuels, plant secondary metabolites or plastic monomers. A reaction network is built with the iterative application of a set of possible enzymes to a set of feed molecules. This network is constantly pruned by applying thermodynamic feasibilty constraints and measures derived from organic chemistry synthesis planning. Furthermore, a variant of Flux Balance Analysis assesses the impact of the predicted pathways on potential host organisms where this pathway should be expressed.
::::work in progress::::
Reconstruction of flux patterns and network structure from isotopomer data
::::work in progress::::
Isotope labeling experiments provide important information that can be used to determine mass fluxes in metabolic network, e.g. where do the carbon atoms of sugar uptake end up in the organism? These mass fluxes can not be measured directly, instead we replace atoms in food molecules with stable isotopes (e.g. 13C). These isotopes end up in different quantities in different metabolites (reporter metabolites). From the distribution of these quantities, we can deduce the mass flux (paths through the reaction network) these atoms must have taken. Usually a fixed network with known stoichiometry is required for this reconstruction, but it is not always available or based on incomplete or faulty data.
The mass flux in a reaction is determined by its atom mapping. By extending that mapping over a reaction network, we can generate hypotheses about the expected isotope distributions. A graph grammar based artificial chemistry provides the means to modify and extend the reaction network on the fly, allowing us to study the impact of perturbations and faulty network data on the predicted flux distributions. Ultimately, it should be possible to predict the network structure from available transformation types (enzymes) and isotopomer data.
::::work in progress::::